bioorthogonal reaction Develoment

Cycloaddition reactions have consumed the field of bioorthogonal chemistry, where highly selective reactions are utilized to study biological systems. In this arena, researchers aim to carry out reactions in vivo without experiencing side-reactions that might interfere with natural processes. It is in this context where cycloadditions shine, avoiding the formation of charged intermediates and providing stable linkages, often in the form of aromatic products. The prototypical "click" reaction—the copper-catalyzed azide–alkyne cycloaddition (CuAAC, X = N)—translates into the biological realm via the introduction of strain (aka pre-distortion) in the strain-promoted azide–alkyne cycloaddition (SPAAC).

From DIFO to SNO-OCTs: Fine-tuning strain and reactivity of cyclooctyne reagents. The strategy of strain activation introduces drawbacks to the stability and selectivity of cyclooctyne reagents. We have introduced an alternate strategy of transition state (TS) stabilization, where electronic effects increase both the stability and reactivity of strained reagents.

We first revealed specific orbital interactions responsible for rate enhancements in electronically activated mono- and difluorocyclooctyne (MOFO and DIFO). The cyclic framework prevents maximal stabilization provided by hyperconjugative π→σ* interactions. We removed cyclic constraints and found that stabilization is maximized by the anti-periplanar arrangement of interacting orbitals. These stereoelectronic interactions provide both assistance to alkyne bending and bond formation.
We then designed an optimal scaffold for stereoelectronic TS stabilization by incorporating endocyclic σ-acceptors into cyclooctynes. These optimized interactions lead to both increased stability and reactivity of strained cyclooctynes. Notably, one endocyclic oxygen provides a larger accelerating effect than two exocyclic fluorines. Various combinations of endo- and exocyclic heteroatoms provide a strategy to tune strain and SPAAC reactivity.

In a collaboration with the Schomaker Group, endocyclic sulfamate-containing cyclooctynes—SNO-OCTs—were synthesized via regioselective aziridination of silyl-substituted allenes. This exciting new class of cyclooctynes both experimentally verify our predictions and reveal a new strategy of remotely controlling alkyne strain via hybridization and stereoelectronic effects. Importantly, these reagents display rates on par with the best of currently available cyclooctyne reagents, yet do not suffer from unwanted side-reactions with endogenous nucleophiles, such as thiolates. These unique features of SNO-OCTs illustrate the advantages of stereoelectronic TS stabilization combined with strain-activation.
Diazo-Selective 1,3-dipolar cycloadditions in the presence of azides. While the strain-promoted azide–alkyne cycloaddition (SPAAC) has overshadowed much of the landscape, the diazo group has overlooked potential in biological applications. 1,3-Dipolar cycloadditions of diazo compounds can be more rapid than those of azides and are more prone to electronic tuning. The ability to survive cellular metabolism and provide unique reactivity makes the diazo group a practical alternative to the azido moiety. Still, shared reactivity with choice strained alkynes (DIBAC, BCN, etc.) illustrates the need to understand cycloaddition reactivity to engender selectivity.
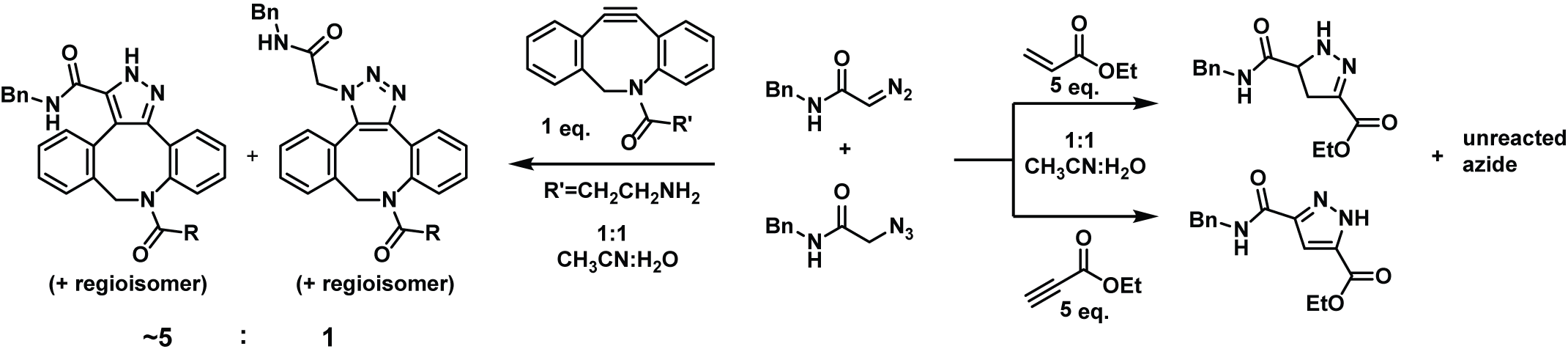
By removing strain and exploring electronic factors controlling reactivity, we have demonstrated diazo-selective reactions with naturally occurring amino acids in the presence of azides at rates comparable to early-generation SPAACs. When pushed to its limits, the strategy of electronic tuning provides cycloadditions with unstrained alkenes and alkynes displaying reaction rates rivaling those of optimized SPAACs. Current efforts are in the pursuit of an optimal balance of strain and electronic effects to provide robust diazo-selective 1,3-dipolar cycloadditions to be carried out in physiological conditions.
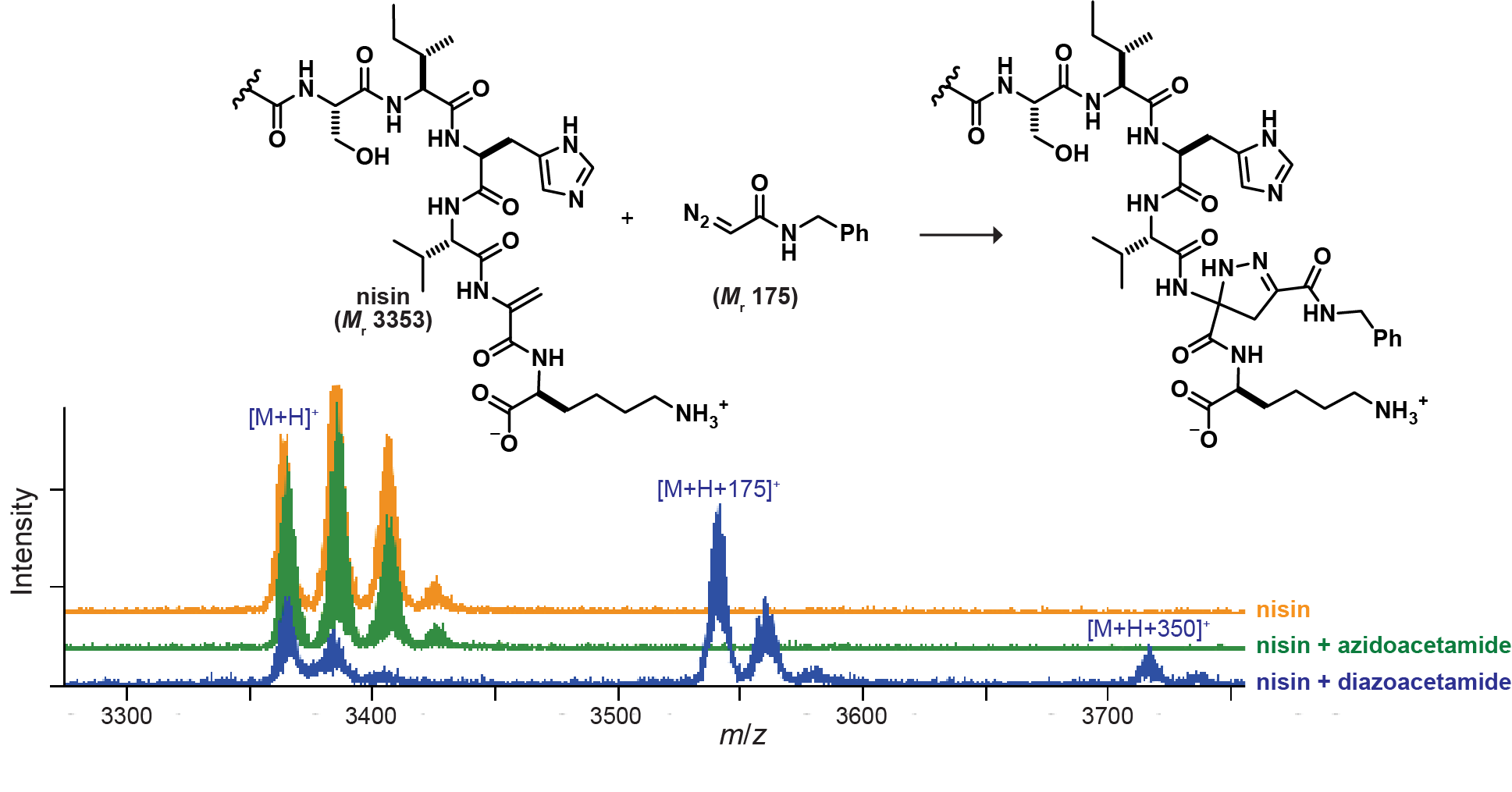

Diazo compounds allow for decreased distortion energies without strain. Most efforts focus on using strain to pre-distort the dipolarophile—which only contributes ~20% of total distortion energies.